Research
Systematic investigation of cancer metabolic reprogramming using multi-omics data
Cancer is intrinsically heterogeneous, spanning multiple spatial and temporal scales. Dissecting this heterogeneity across resolutions remains challenging. Our overarching strategy is to integrate state-of-the-art sequencing technologies with rigorous computational modeling to characterize tumor metabolic programs and their interplay with the tumor microenvironment.
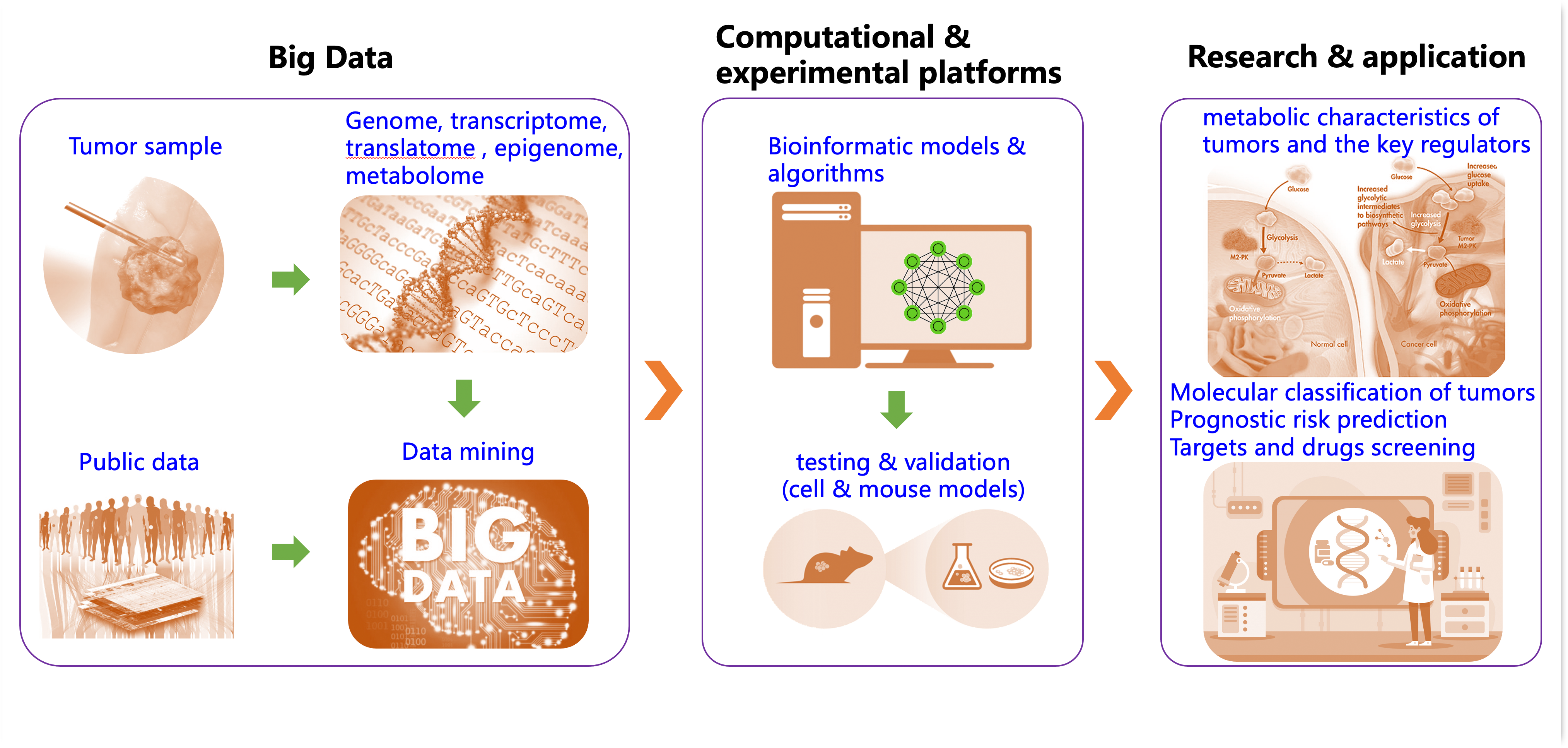
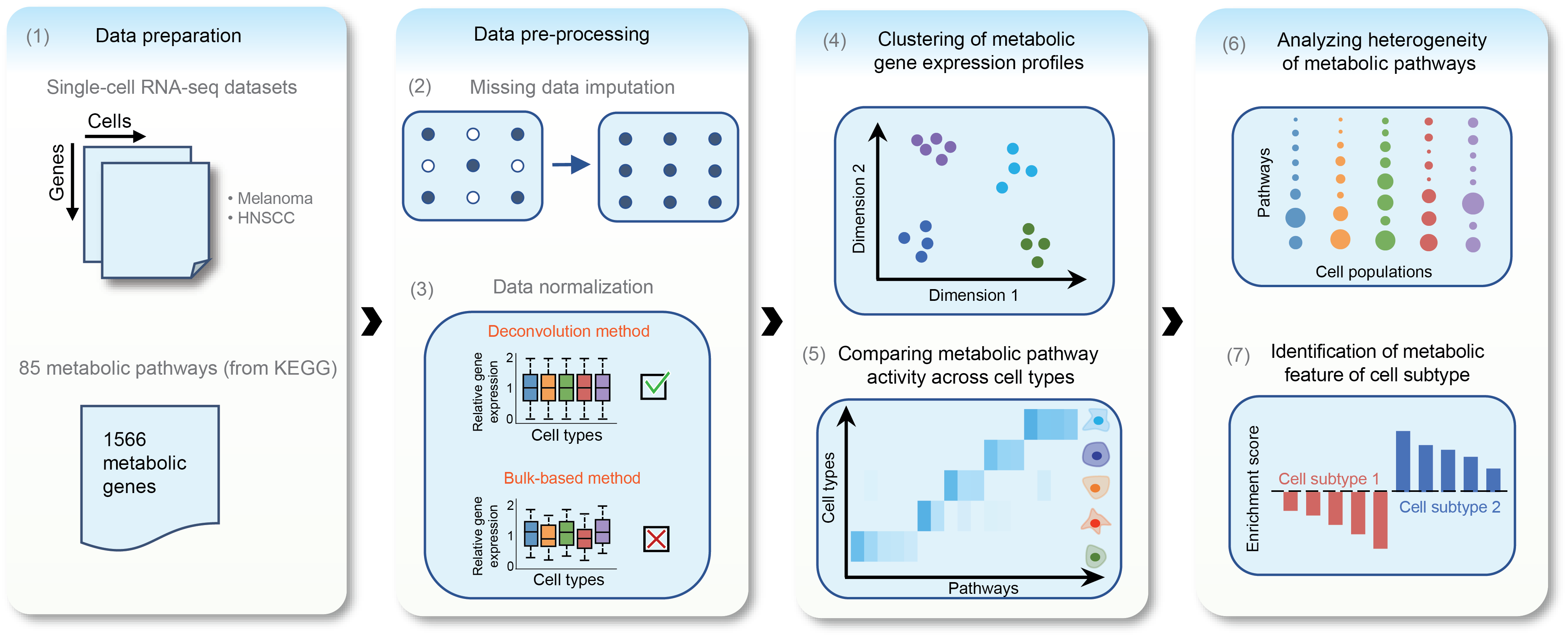
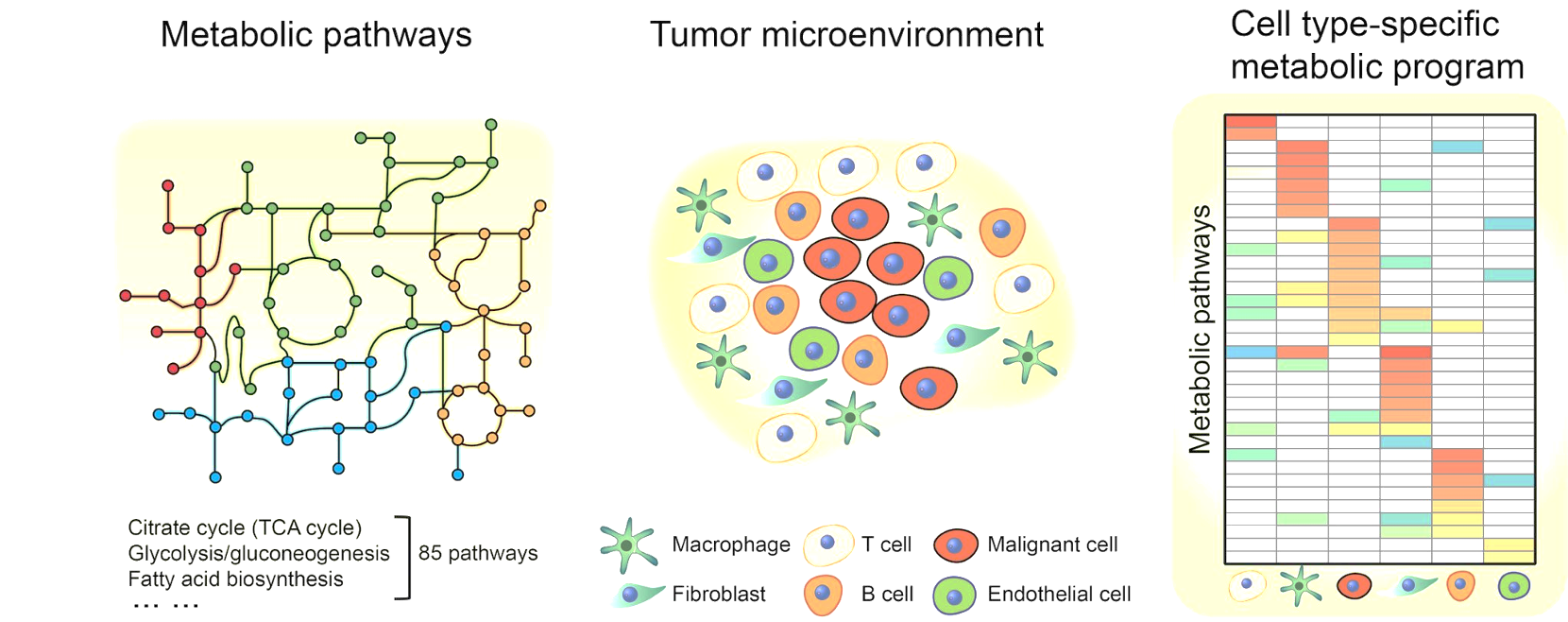
We developed computational algorithms and workflows for profiling metabolic programs at single-cell resolution (https://github.com/zhengtaoxiao/Single-Cell-Metabolic-Landscape). Applying this framework to two representative human cancers—melanoma and head and neck cancer—we systematically mapped metabolic features across cell types and subtypes, and derived organizing principles of tumor–microenvironment interactions. (Xiao et al., 2019, Nat. Commun. )
This study has been selected as top10 cancer research publication by the Board of the European Association for Cancer Research (https://magazine.eacr.org/the-eacrs-top-10-cancer-research-publications-november-2019/8/).
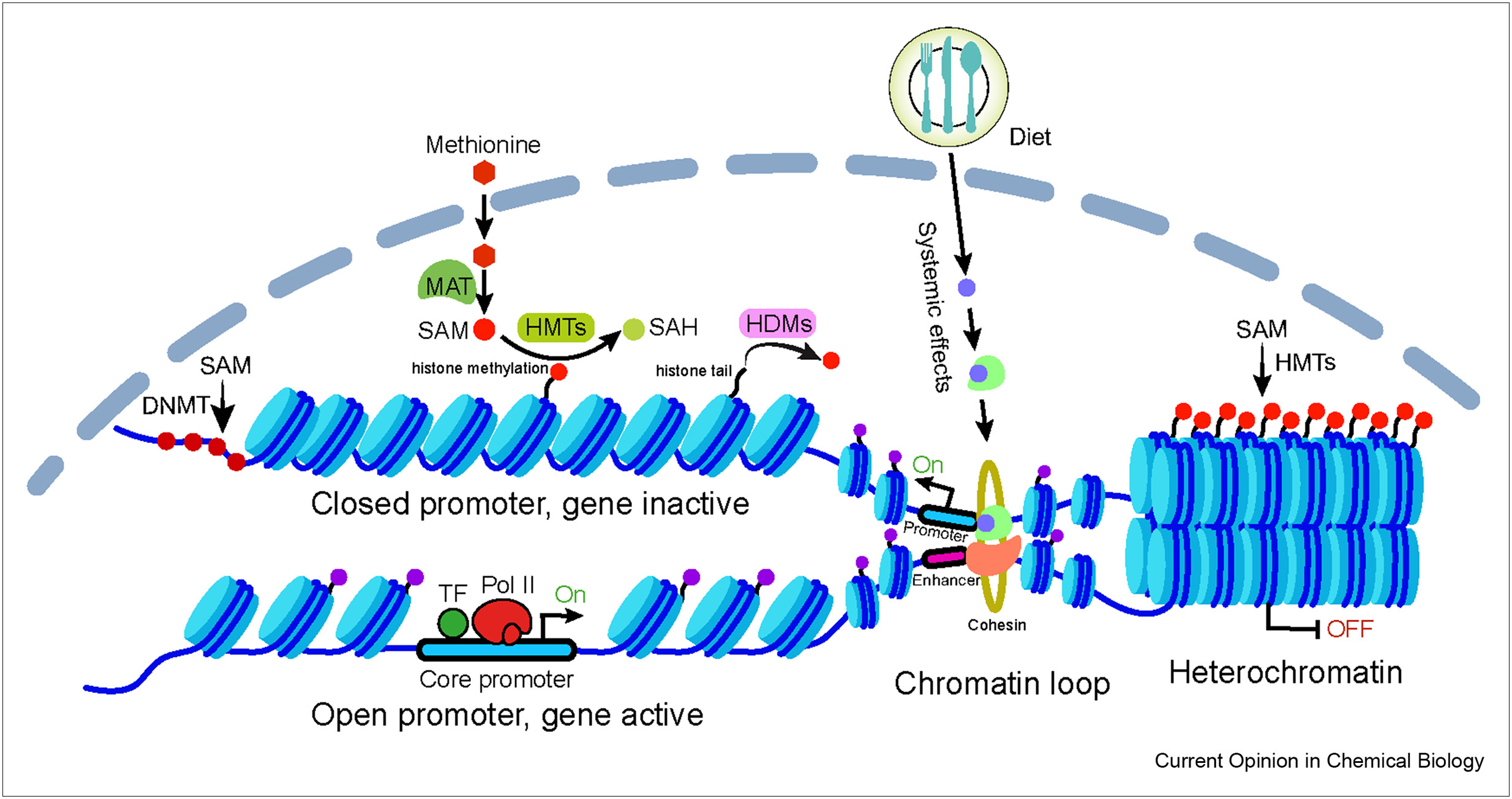
Metabolic effects on chromatin modifications and structures
Chromatin and associated epigenetic marks provide important platforms for gene regulation in response to metabolic changes associated with environmental exposures, including physiological stress, nutritional deprivation, and starvation. The fluctuations of key metabolites can influence chromatin modifications, but their effects on chromatin structure (e.g. chromatin compaction, nucleosome arrangement, and chromatin loops) and how they appropriately deposit specific chemical modification on chromatin are largely unknown. We are seeking to investigate the metabolic effects on chromatin modifications and structure, as well as consequences on gene regulation (Xiao & Locasale 2021. Current Opinion in Chemical Biology).
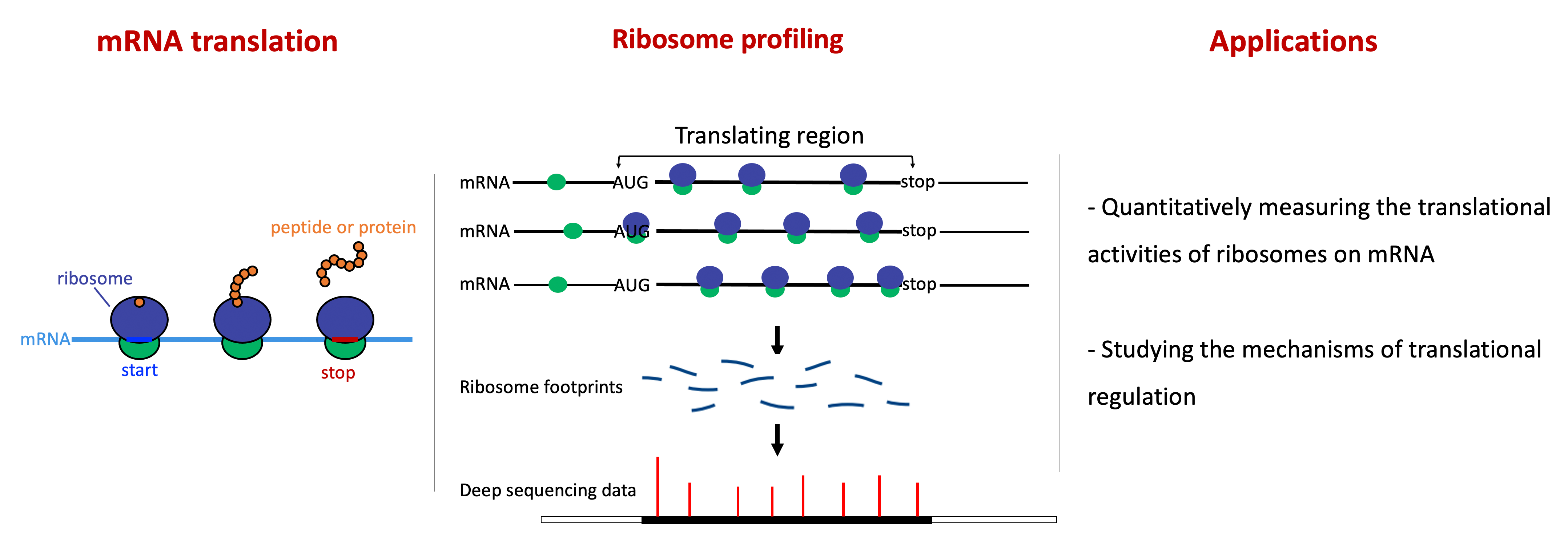
Identification of genome-wide translation dysregulations and novel translated peptides with ribosome profiling data
By capturing and sequencing the RNA fragments protected by translating ribosomes, ribosome profiling provides snapshots of translation at subcodon resolution. Specialized for the ribosome profiling data, we developped a computational piple for comprehensively and accurately identifing differentially translated genes in pairwise comparisons (Xiao et al. 2016. Nat. Commun.) and another piple for de novo annotating and charaterizing the translatome (Xiao et al. 2018. NAR.). We provided step-by-step instructions for identifying the actively translated open reading frames (ORFs) and evaluating the translation rates of the predicted ORFs with ribosome profiling data (Zhu et al. 2022. JoVE).