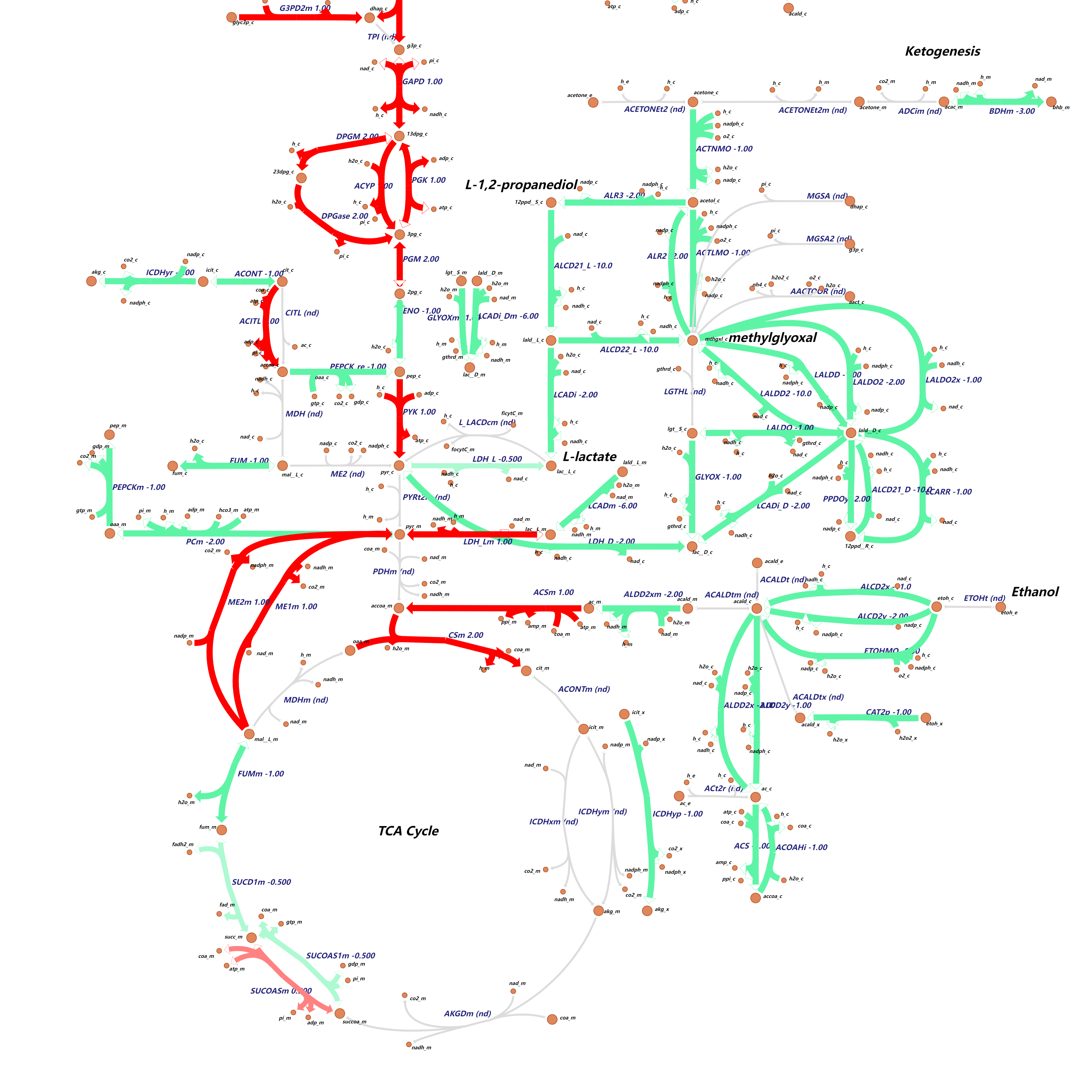
Metabolic peculiarities of cancer cells can be traced to abnormal variations in the activities of particular metabolic enzymes controlling the corresponding metabolic processes. Accurately calculating metabolic enzyme activity is highly desired for exploiting metabolic vulnerabilities in cancers. To reach this goal, we are developping a strategy for integrating RNA-seq data with metabolic networks to predicting the activities of key metabolic enzymes. Our method gives a good prediction of metabolic behavior of different tumors. On this ground, we will further leverage cutting-edge bioinformatic approaches to investigate the various facets of cancer metabolism.
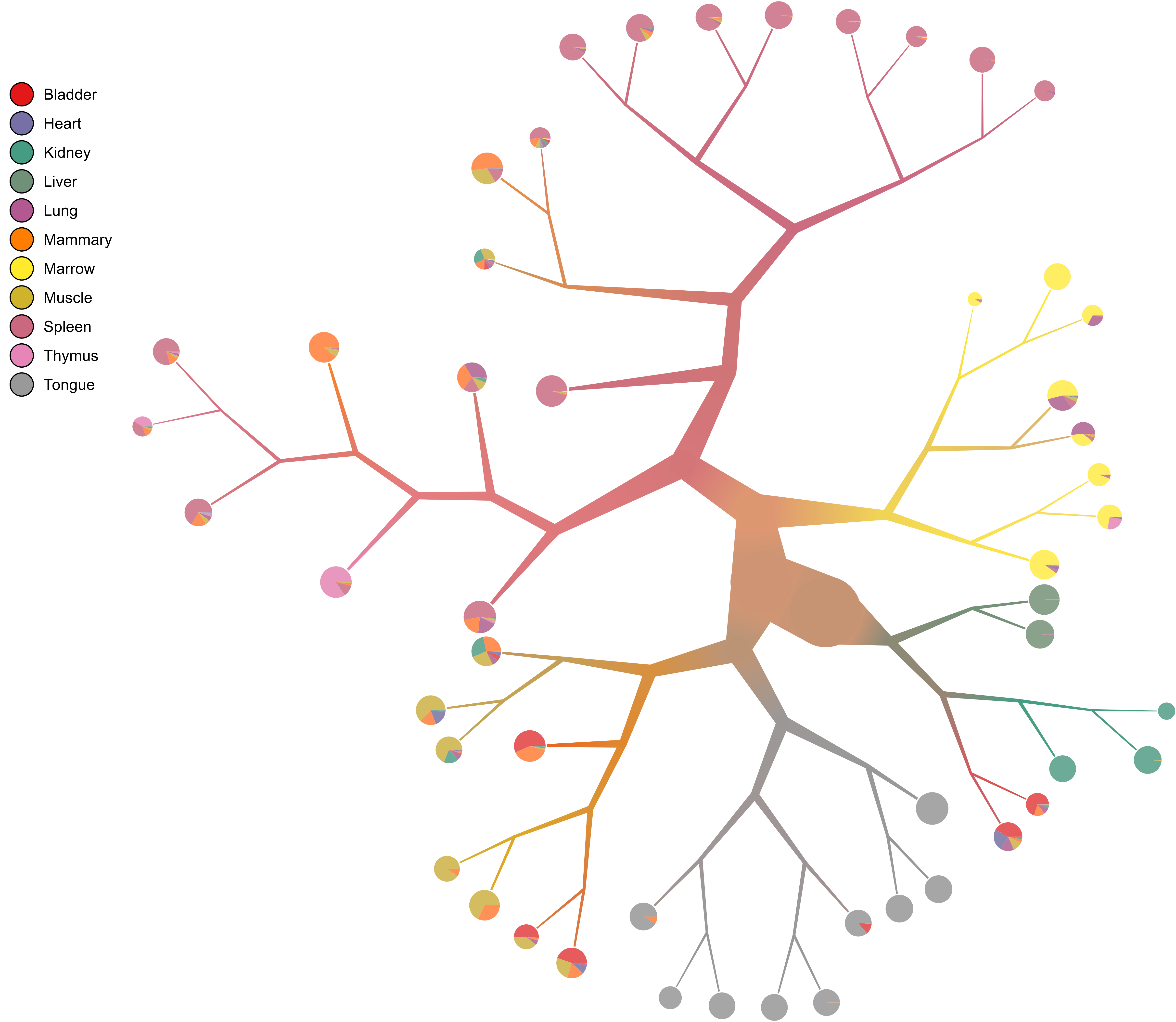
Cell clustering is a key step in scRNA-seq data analysis, which is being challenged by high dimension, significant dropout rate, and hierarchies of nested cell clusters in scRNA-seq data. We aim to develop a novel algorithm based on the cluster condensation strategy for recursively clustering cell populations using scRNA-seq transcriptome. We also intend to define the hierarchical relationship of cell clusters in the tumor microenvironment.
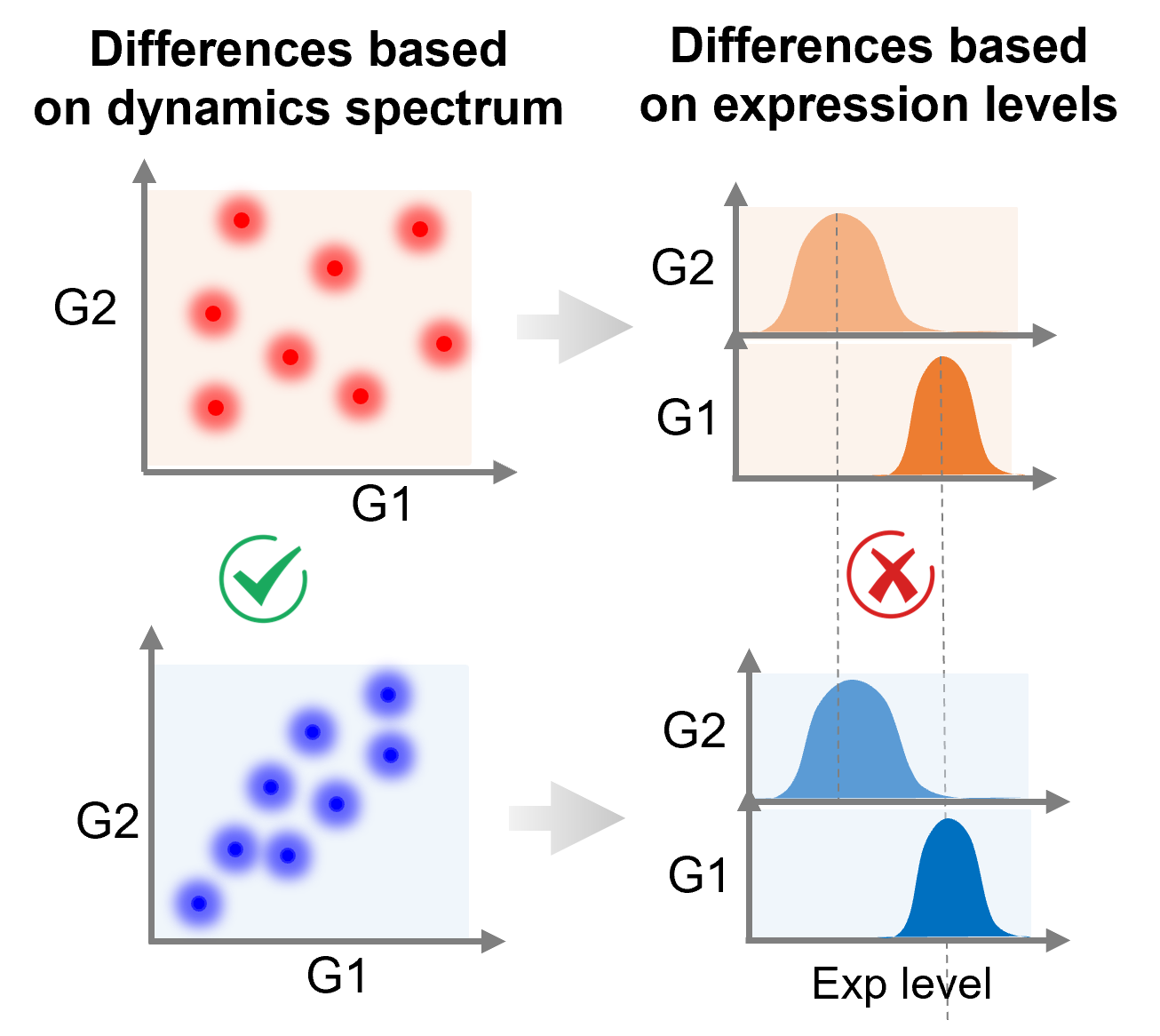
Identification of cell identity and key genes that drive cell transition at single-cell resolution is important for dissecting the tumor microenvironment and understanding the cause of the disease such as cancer. Tranditionally used methods for this purpose is differential gene expression analysis. However, the performances of this kind of methods are highly dependent on the detected expression of individual genes, which overlook the post-transcriptional regulation and the interactions among those genes. To overcome this challenge, we are developping a computational model for examing the dynamic trends of gene-gene interactions and its association with cell function. The benchmarking results show that our method has a great performance for identify the key genes and gene regulation that participate in cell-type-specific functions.